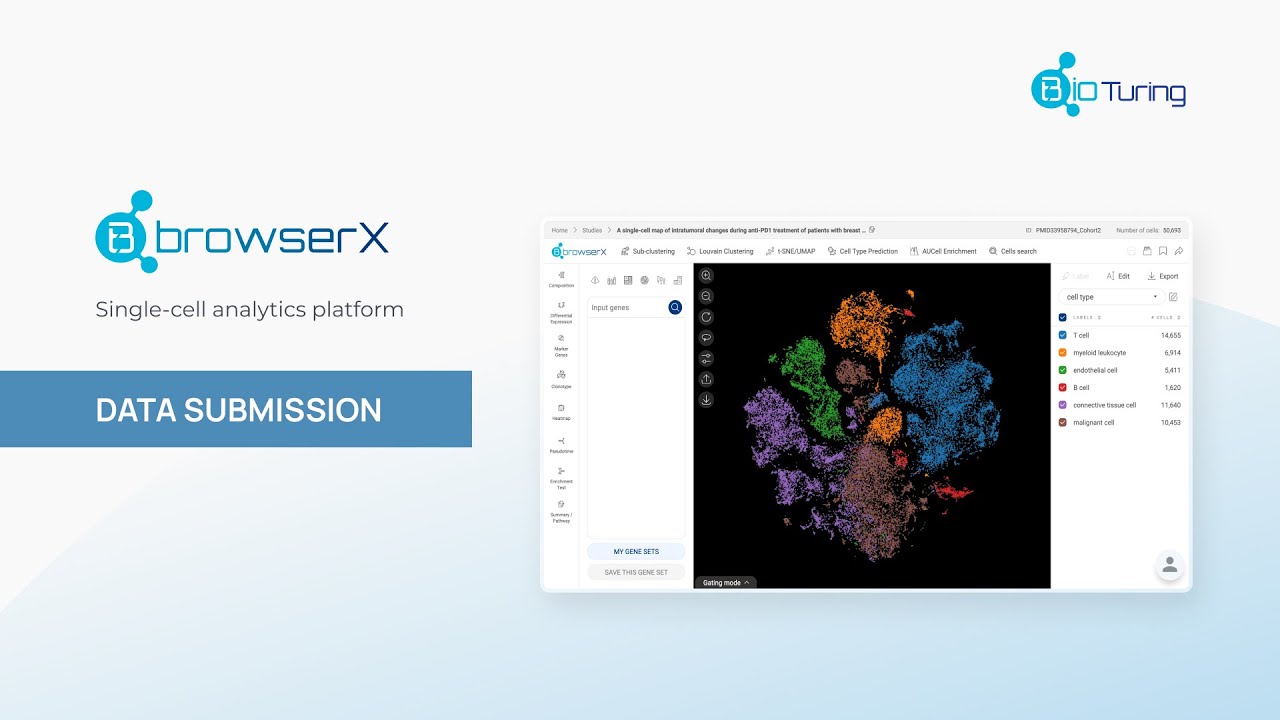
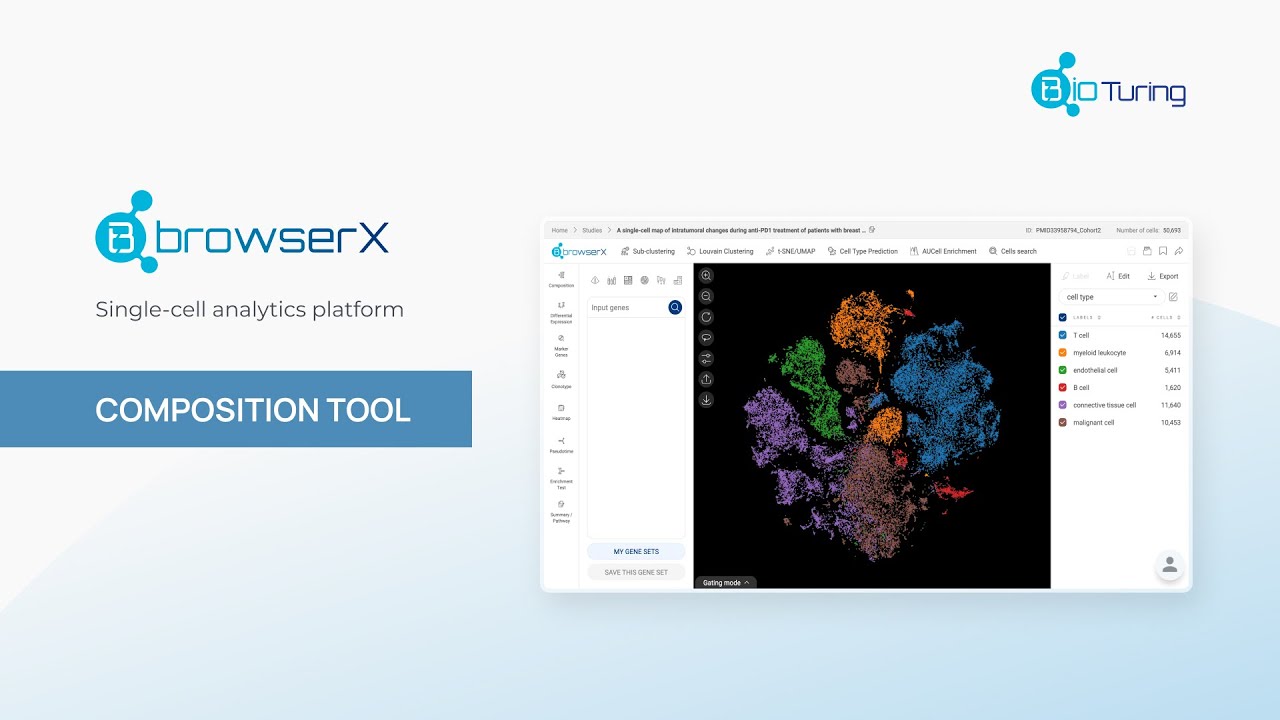
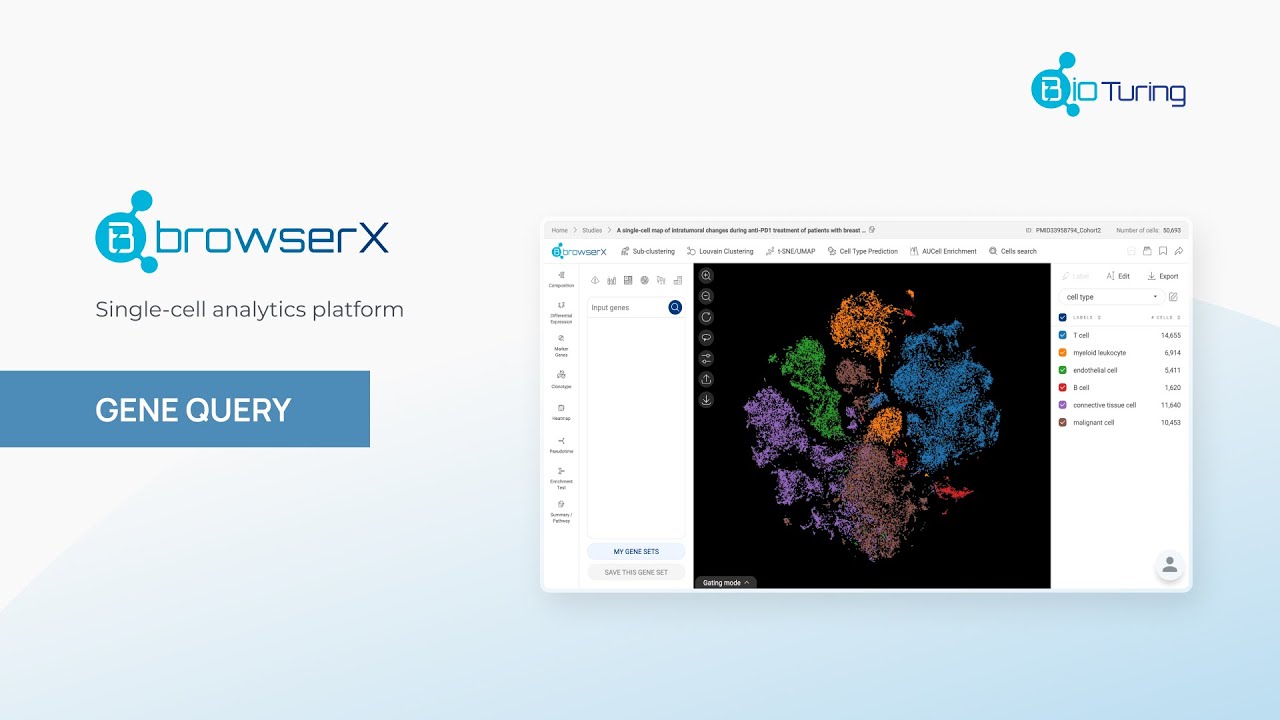
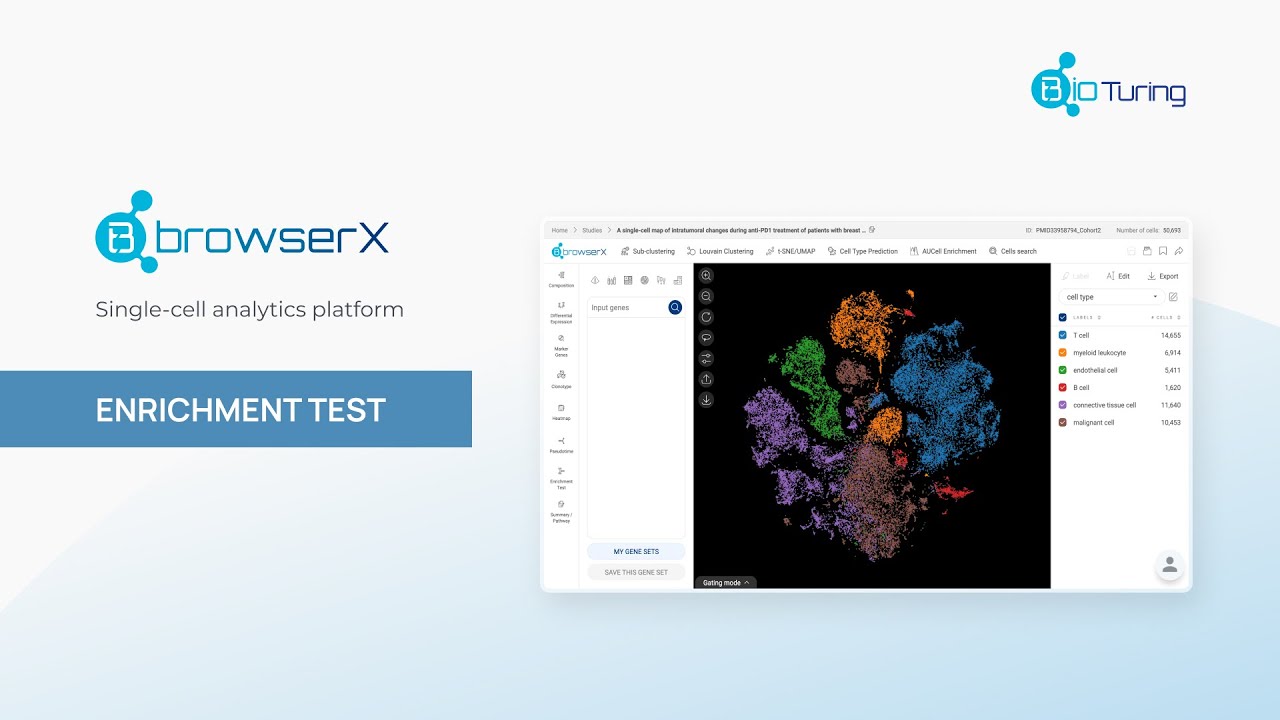
Methods
IC-VAE: A Novel Deep Learning Framework for Interpreting Multiplexed
Tissue Imaging Data.
Interpreting protein expression in multiplexed tissue imaging data
presents a significant challenge due to the high dimensionality of
the resulting images, the variety of intracellular structures, cell
shapes resulting from 2-D tissue sectioning, and the presence of
technological noise and imaging artifacts. Here, we introduce the
Information-Controlled Variational Autoencoder (IC-VAE), a deep
generative model designed to tackle this challenge. The contribution
of IC-VAE to the VAE framework is the ability to control the shared
information among latent subspaces. We use IC-VAE to factorize each
cell's image into its true protein expression, various cellular
components, and background noise, while controlling the shared
information among some of these components. Compared with other
normalization methods, this approach leads to superior results in
downstream analysis, such as analyzing the expression of biomarkers,
classification for cell types, or visualizing cell clusters using
t-SNE/UMAP techniques.
bioRxiv 2023.11.06.565771; doi: 10.1101/2023.11.06.565771v1
Venice: A New Algorithm for Finding Marker Genes in Single-Cell
Transcriptomic Data
Most widely used tools for finding marker genes in single cell data
(SeuratT/NegBinom/Poisson, CellRanger, EdgeR, limmatrend) use a
conventional definition of differentially expressed genes: genes
with different mean expression values. However, in single-cell data,
a cell population can be a mixture of many cell types/cell states,
hence the mean expression of genes cannot represent the whole
population. In addition, these tools assume that gene expression of
a population belongs to a specific family of distribution. This
assumption is often violated in single-cell data. In this work, we
define marker genes of a cell population as genes that can be used
to distinguish cells in the population from cells in other
populations. Besides log-fold change, we devise a new metric to
classify genes into up-regulated, down-regulated, and transitional
states. In a benchmark for finding up-regulated and down-regulated
genes, our tool outperforms all compared methods, including Seurat,
ROTS, scDD, edgeR, MAST, limma, normal t-test, Wilcoxon and
Kolmogorov–Smirnov test. Our method is much faster than all compared
methods, therefore, enables interactive analysis for large
single-cell data sets in BioTuring Browser. Venice algorithm is
available within Signac package:
https://github.com/bioturing/signac).
bioRxiv 2020.11.16.384479; doi: 10.1101/2020.11.16.384479v1
An Entropy Approach for Choosing Gene Expression Cutoff
Annotating cell types using single-cell transcriptome data usually
requires binarizing the expression data to distinguish between the
background noise vs. real expression or low expression vs. high
expression cases. A common approach is choosing a “reasonable”
cutoff value, but it remains unclear how to choose it. In this work,
we describe a simple yet effective approach for finding this
threshold value.
A common procedure to annotate cell types in a single-cell RNA-seq study is to first perform graph-based clustering, and further check the expression of some marker genes in each cluster. In some cases, scientists need to distinguish between the real expression of a gene vs the background expression. In other cases, they need to know if a gene expresses highly in one cluster and lowly in another cluster (e.g., NK CD56bright and NK CD56dim). This requires choosing a threshold to binarize the expression data. It remains unclear how to choose this threshold. Here, we propose to binarize the data in a way that minimizes the clustering information loss. Below, we describe the formulation in detail.
bioRxiv 2022.05.05.490711; doi: 10.1101/2022.05.05.490711v1
A common procedure to annotate cell types in a single-cell RNA-seq study is to first perform graph-based clustering, and further check the expression of some marker genes in each cluster. In some cases, scientists need to distinguish between the real expression of a gene vs the background expression. In other cases, they need to know if a gene expresses highly in one cluster and lowly in another cluster (e.g., NK CD56bright and NK CD56dim). This requires choosing a threshold to binarize the expression data. It remains unclear how to choose this threshold. Here, we propose to binarize the data in a way that minimizes the clustering information loss. Below, we describe the formulation in detail.
Hera-T: an efficient and accurate approach for quantifying gene
abundances from 10X-Chromium data with high rates of non-exonic
reads
An important but rarely discussed phenomenon in single cell data
generated by the 10X-Chromium protocol is that the fraction of
non-exonic reads is very high. This number usually exceeds 30% of
the total reads. Without aligning them to a complete genome
reference, non-exonic reads can be erroneously aligned to the
transcriptome reference with higher error rates. To tackle this
problem, Cell Ranger chooses to firstly align reads against the
whole genome, and at a later step, uses a genome annotation to
select reads that align to the transcriptome. Despite its high
running time and large memory consumption, Cell Ranger remains the
most widely used tool to quantify 10XGenomics single cell RNA-Seq
data for its accuracy.
In this work, we introduce Hera-T, a fast and accurate tool for estimating gene abundances in single cell data generated by the 10X-Chromium protocol. By devising a new strategy for aligning reads to both transcriptome and genome references, Hera-T reduces both running time and memory consumption from 10 to 100 folds while giving similar results compared to Cell Ranger’s. Hera-T also addresses some difficult splicing alignment scenarios that Cell Ranger fails to address, and therefore, obtains better accuracy compared to Cell Ranger. Excluding the reads in those scenarios, Hera-T and Cell Ranger results have correlation scores > 0.99.
For a single-cell data set with 49 million of reads, Cell Ranger took 3 hours (179 minutes) while Hera-T took 1.75 minutes; for another single-cell data set with 784 millions of reads, Cell Ranger took about 25 hours while Hera-T took 32 minutes. For those data sets, Cell Ranger completely used all 32 GB of memory while Hera-T consumed at most 8 GB. Hera-T package is available for download at: https://bioturing.com/product/hera-t
bioRxiv 530501; doi: 10.1101/530501
In this work, we introduce Hera-T, a fast and accurate tool for estimating gene abundances in single cell data generated by the 10X-Chromium protocol. By devising a new strategy for aligning reads to both transcriptome and genome references, Hera-T reduces both running time and memory consumption from 10 to 100 folds while giving similar results compared to Cell Ranger’s. Hera-T also addresses some difficult splicing alignment scenarios that Cell Ranger fails to address, and therefore, obtains better accuracy compared to Cell Ranger. Excluding the reads in those scenarios, Hera-T and Cell Ranger results have correlation scores > 0.99.
For a single-cell data set with 49 million of reads, Cell Ranger took 3 hours (179 minutes) while Hera-T took 1.75 minutes; for another single-cell data set with 784 millions of reads, Cell Ranger took about 25 hours while Hera-T took 32 minutes. For those data sets, Cell Ranger completely used all 32 GB of memory while Hera-T consumed at most 8 GB. Hera-T package is available for download at: https://bioturing.com/product/hera-t
A revisit of RSEM generative model and its EM algorithm for
quantifying transcript abundances
RSEM has been mainly known for its accuracy in transcript abundance
quantification. However, its quantification time is extremely high
compared to that of recent quantification tools. In this paper, we
revised the RSEM’s EM algorithm. In particular, we derived accurate
M-step updates to eliminate incorrect heuristic updates in RSEM. We
also implement some optimizations that reduce the quantification
time about a hundred times while still have better accuracy compared
to RSEM. In particular, we noticed that different parameters have
different convergence rates, therefore we identified and removed
early converged parameters to significantly reduce the model
complexity in further iterations, and we also use SQUAREM method to
further speed up the convergence rate. We implemented these
revisions in a packaged named Hera-EM, with source code available
at: https://github.com/bioturing/hera/tree/master/hera-EM
bioRxiv 503672; doi: 10.1101/503672
SPAdes: a new genome assembly algorithm and its applications to
single-cell sequencing
The lion's share of bacteria in various environments cannot be
cloned in the laboratory and thus cannot be sequenced using existing
technologies. A major goal of single-cell genomics is to complement
gene-centric metagenomic data with whole-genome assemblies of
uncultivated organisms. Assembly of single-cell data is challenging
because of highly non-uniform read coverage as well as elevated
levels of sequencing errors and chimeric reads. We describe SPAdes,
a new assembler for both single-cell and standard (multicell)
assembly, and demonstrate that it improves on the recently released
E+V−SC assembler (specialized for single-cell data) and on popular
assemblers Velvet and SoapDeNovo (for multicell data). SPAdes
generates single-cell assemblies, providing information about
genomes of uncultivatable bacteria that vastly exceeds what may be
obtained via traditional metagenomics studies. SPAdes is available
online (http://bioinf.spbau.ru/spades). It is distributed as open
source software.
J Comput Biol 2012 May;19(5):455-77; doi: 10.1089/cmb.2012.0021
Differential responses to lithium in hyperexcitable neurons from
patients with bipolar disorder
The lion's share of bacteria in various environments cannot be
cloned in the laboratory and thus cannot be sequenced using existing
technologies. A major goal of single-cell genomics is to complement
gene-centric metagenomic data with whole-genome assemblies of
uncultivated organisms. Assembly of single-cell data is challenging
because of highly non-uniform read coverage as well as elevated
levels of sequencing errors and chimeric reads. We describe SPAdes,
a new assembler for both single-cell and standard (multicell)
assembly, and demonstrate that it improves on the recently released
E+V−SC assembler (specialized for single-cell data) and on popular
assemblers Velvet and SoapDeNovo (for multicell data). SPAdes
generates single-cell assemblies, providing information about
genomes of uncultivatable bacteria that vastly exceeds what may be
obtained via traditional metagenomics studies. SPAdes is available
online (http://bioinf.spbau.ru/spades). It is distributed as open
source software.
Nature. 2016 Feb 11;530(7589):242; doi: 10.1038/nature1618
Using single nuclei for RNA-seq to capture the transcriptome of
postmortem neurons
A protocol is described for sequencing the transcriptome of a cell
nucleus. Nuclei are isolated from specimens and sorted by FACS, cDNA
libraries are constructed and RNA-seq is performed, followed by data
analysis. Some steps follow published methods (Smart-seq2 for cDNA
synthesis and Nextera XT barcoded library preparation) and are not
described in detail here. Previous single-cell approaches for
RNA-seq from tissues include cell dissociation using protease
treatment at 30 °C, which is known to alter the transcriptome. We
isolate nuclei at 4 °C from tissue homogenates, which cause minimal
damage. Nuclear transcriptomes can be obtained from postmortem human
brain tissue stored at -80 °C, making brain archives accessible for
RNA-seq from individual neurons. The method also allows
investigation of biological features unique to nuclei, such as
enrichment of certain transcripts and precursors of some noncoding
RNAs. By following this procedure, it takes about 4 d to construct
cDNA libraries that are ready for sequencing.
Nat Protoc. 2016 Mar;11(3):499-524; doi: 10.1038/nprot.2016.015
Ragout—a reference-assisted assembly tool for bacterial genomes
Bacterial genomes are simpler than mammalian ones, and yet
assembling the former from the data currently generated by
high-throughput short-read sequencing machines still results in
hundreds of contigs. To improve assembly quality, recent studies
have utilized longer Pacific Biosciences (PacBio) reads or jumping
libraries to connect contigs into larger scaffolds or help
assemblers resolve ambiguities in repetitive regions of the genome.
However, their popularity in contemporary genomic research is still
limited by high cost and error rates. In this work, we explore the
possibility of improving assemblies by using complete genomes from
closely related species/strains. We present Ragout, a genome
rearrangement approach, to address this problem. In contrast with
most reference-guided algorithms, where only one reference genome is
used, Ragout uses multiple references along with the evolutionary
relationship among these references in order to determine the
correct order of the contigs. Additionally, Ragout uses the assembly
graph and multi-scale synteny blocks to reduce assembly gaps caused
by small contigs from the input assembly. In simulations as well as
real datasets, we believe that for common bacterial species, where
many complete genome sequences from related strains have been
available, the current high-throughput short-read sequencing
paradigm is sufficient to obtain a single high-quality scaffold for
each chromosome.
Bioinformatics. 2014 Jun 15;30(12):i302-9; doi:
10.1093/bioinformatics/btu280
Sixteen diverse laboratory mouse reference genomes define
strain-specific haplotypes and novel functional loci
We report full-length draft de novo genome assemblies for 16 widely
used inbred mouse strains and find extensive strain-specific
haplotype variation. We identify and characterize 2,567 regions on
the current mouse reference genome exhibiting the greatest sequence
diversity. These regions are enriched for genes involved in pathogen
defence and immunity and exhibit enrichment of transposable elements
and signatures of recent retrotransposition events. Combinations of
alleles and genes unique to an individual strain are commonly
observed at these loci, reflecting distinct strain phenotypes. We
used these genomes to improve the mouse reference genome, resulting
in the completion of 10 new gene structures. Also, 62 new coding
loci were added to the reference genome annotation. These genomes
identified a large, previously unannotated, gene (Efcab3-like)
encoding 5,874 amino acids. Mutant Efcab3-like mice display
anomalies in multiple brain regions, suggesting a possible role for
this gene in the regulation of brain development.
Nat Genet. 2018 Nov;50(11):1574-1583; doi: 10.1038/s41588-018-0223-8
Sibelia: a scalable and comprehensive synteny block generation tool
for closely related microbial genomes
Comparing strains within the same microbial species has proven
effective in the identification of genes and genomic regions
responsible for virulence, as well as in the diagnosis and treatment
of infectious diseases. In this paper, we present Sibelia, a tool
for finding synteny blocks in multiple closely related microbial
genomes using iterative de Bruijn graphs. Unlike most other tools,
Sibelia can find synteny blocks that are repeated within genomes as
well as blocks shared by multiple genomes. It represents synteny
blocks in a hierarchy structure with multiple layers, each of which
representing a different granularity level. Sibelia has been
designed to work efficiently with a large number of microbial
genomes; it finds synteny blocks in 31 S. aureus genomes within 31
minutes and in 59 E.coli genomes within 107 minutes on a standard
desktop. Sibelia software is distributed under the GNU GPL v2
license and is available at: https://github.com/bioinf/Sibelia.
Sibelia’s web-server is available at:
http://etool.me/software/sibelia.
Lecture Notes in Computer Science(), vol 8126. Springer, Berlin,
Heidelberg; doi: 10.1007/978-3-642-40453-5_17
Paired de bruijn graphs: a novel approach for incorporating mate
pair information into genome assemblers
The recent proliferation of next generation sequencing with short
reads has enabled many new experimental opportunities but, at the
same time, has raised formidable computational challenges in genome
assembly. One of the key advances that has led to an improvement in
contig lengths has been mate pairs, which facilitate the assembly of
repeating regions. Mate pairs have been algorithmically incorporated
into most next generation assemblers as various heuristic
post-processing steps to correct the assembly graph or to link
contigs into scaffolds. Such methods have allowed the identification
of longer contigs than would be possible with single reads; however,
they can still fail to resolve complex repeats. Thus, improved
methods for incorporating mate pairs will have a strong effect on
contig length in the future. Here, we introduce the paired de Bruijn
graph, a generalization of the de Bruijn graph that incorporates
mate pair information into the graph structure itself instead of
analyzing mate pairs at a post-processing step. This graph has the
potential to be used in place of the de Bruijn graph in any de
Bruijn graph based assembler, maintaining all other assembly steps
such as error-correction and repeat resolution. Through assembly
results on simulated perfect data, we argue that this can
effectively improve the contig sizes in assembly.
J Comput Biol. 2011 Nov; 18(11): 1625–1634; doi:
10.1089/cmb.2011.0151
ExSPAnder: a universal repeat resolver for DNA fragment assembly
Next-generation sequencing (NGS) technologies have raised a
challenging de novo genome assembly problem that is further
amplified in recently emerged single-cell sequencing projects. While
various NGS assemblers can use information from several libraries of
read-pairs, most of them were originally developed for a single
library and do not fully benefit from multiple libraries. Moreover,
most assemblers assume uniform read coverage, condition that does
not hold for single-cell projects where utilization of read-pairs is
even more challenging. We have developed an exSPAnder algorithm that
accurately resolves repeats in the case of both single and multiple
libraries of read-pairs in both standard and single-cell assembly
projects.
Bioinformatics, Volume 30, Issue 12, 15 June 2014, Pages i293–i301;
doi: 10.1093/bioinformatics/btu266
DRIMM-Synteny: decomposing genomes into evolutionary conserved
segments
Motivation: The rapidly increasing set of sequenced genomes
highlights the importance of identifying the synteny blocks in
multiple and/or highly duplicated genomes. Most synteny block
reconstruction algorithms use genes shared over all genomes to
construct the synteny blocks for multiple genomes. However, the
number of genes shared among all genomes quickly decreases with the
increase in the number of genomes.
Results: We propose the Duplications and Rearrangements In Multiple Mammals (DRIMM)-Synteny algorithm to address this bottleneck and apply it to analyzing genomic architectures of yeast, plant and mammalian genomes. We further combine synteny block generation with rearrangement analysis to reconstruct the ancestral preduplicated yeast genome.
Bioinformatics. 2010 Oct 15;26(20):2509-16. doi; doi:
10.1093/bioinformatics/btq465
Results: We propose the Duplications and Rearrangements In Multiple Mammals (DRIMM)-Synteny algorithm to address this bottleneck and apply it to analyzing genomic architectures of yeast, plant and mammalian genomes. We further combine synteny block generation with rearrangement analysis to reconstruct the ancestral preduplicated yeast genome.
Mitochondrial aging defects emerge in directly reprogrammed human
neurons due to their metabolic profile
Mitochondria are a major target for aging and are instrumental in
the age-dependent deterioration of the human brain, but studying
mitochondria in aging human neurons has been challenging. Direct
fibroblast-to-induced neuron (iN) conversion yields functional
neurons that retain important signs of aging, in contrast to iPSC
differentiation. Here, we analyzed mitochondrial features in iNs
from individuals of different ages. iNs from old donors display
decreased oxidative phosphorylation (OXPHOS)-related gene
expression, impaired axonal mitochondrial morphologies, lower
mitochondrial membrane potentials, reduced energy production, and
increased oxidized proteins levels. In contrast, the fibroblasts
from which iNs were generated show only mild age-dependent changes,
consistent with a metabolic shift from glycolysis-dependent
fibroblasts to OXPHOS-dependent iNs. Indeed, OXPHOS-induced old
fibroblasts show increased mitochondrial aging features similar to
iNs. Our data indicate that iNs are a valuable tool for studying
mitochondrial aging and support a bioenergetic explanation for the
high susceptibility of the brain to aging.
Cell Rep. 2018 May 29;23(9):2550-2558; doi:
10.1016/j.celrep.2018.04.105
TwoPaCo: an efficient algorithm to build the compacted de Bruijn
graph from many complete genomes
Motivation: de Bruijn graphs have been proposed as a data structure
to facilitate the analysis of related whole genome sequences, in
both a population and comparative genomic settings. However, current
approaches do not scale well to many genomes of large size (such as
mammalian genomes).
Results: In this article, we present TWOPACO, a simple and scalable low memory algorithm for the direct construction of the compacted de Bruijn graph from a set of complete genomes. We demonstrate that it can construct the graph for 100 simulated human genomes in less than a day and eight real primates in < 2 h, on a typical shared-memory machine. We believe that this progress will enable novel biological analyses of hundreds of mammalian-sized genomes.
Availability and Implementation: Our code and data is available for download from github.com/medvedevgroup/TwoPaCo.
Bioinformatics, Volume 33, Issue 24, 15 December 2017, Pages
4024–4032; doi: 10.1093/bioinformatics/btw609
Results: In this article, we present TWOPACO, a simple and scalable low memory algorithm for the direct construction of the compacted de Bruijn graph from a set of complete genomes. We demonstrate that it can construct the graph for 100 simulated human genomes in less than a day and eight real primates in < 2 h, on a typical shared-memory machine. We believe that this progress will enable novel biological analyses of hundreds of mammalian-sized genomes.
Availability and Implementation: Our code and data is available for download from github.com/medvedevgroup/TwoPaCo.
Repeat associated mechanisms of genome evolution and function
revealed by the Mus caroli and Mus pahari genomes
Understanding the mechanisms driving lineage-specific evolution in
both primates and rodents has been hindered by the lack of sister
clades with a similar phylogenetic structure having high-quality
genome assemblies. Here, we have created chromosome-level assemblies
of the Mus caroli and Mus pahari genomes. Together with the Mus
musculus and Rattus norvegicus genomes, this set of rodent genomes
is similar in divergence times to the Hominidae
(human-chimpanzee-gorilla-orangutan). By comparing the evolutionary
dynamics between the Muridae and Hominidae, we identified punctate
events of chromosome reshuffling that shaped the ancestral karyotype
of Mus musculus and Mus caroli between 3 and 6 million yr ago, but
that are absent in the Hominidae. Hominidae show between four- and
sevenfold lower rates of nucleotide change and feature turnover in
both neutral and functional sequences, suggesting an underlying
coherence to the Muridae acceleration. Our system of matched,
high-quality genome assemblies revealed how specific classes of
repeats can play lineage-specific roles in related species. Recent
LINE activity has remodeled protein-coding loci to a greater extent
across the Muridae than the Hominidae, with functional consequences
at the species level such as reproductive isolation. Furthermore, we
charted a Muridae-specific retrotransposon expansion at
unprecedented resolution, revealing how a single nucleotide mutation
transformed a specific SINE element into an active CTCF binding site
carrier specifically in Mus caroli, which resulted in thousands of
novel, species-specific CTCF binding sites. Our results show that
the comparison of matched phylogenetic sets of genomes will be an
increasingly powerful strategy for understanding mammalian biology.
Genome Res. 2018 Apr; 28(4): 448–459; doi: 10.1101/gr.234096.117
Chromosome assembly of large and complex genomes using multiple
references
Despite the rapid development of sequencing technologies, the
assembly of mammalian-scale genomes into complete chromosomes
remains one of the most challenging problems in bioinformatics. To
help address this difficulty, we developed Ragout 2, a
reference-assisted assembly tool that works for large and complex
genomes. By taking one or more target assemblies (generated from an
NGS assembler) and one or multiple related reference genomes, Ragout
2 infers the evolutionary relationships between the genomes and
builds the final assemblies using a genome rearrangement approach.
By using Ragout 2, we transformed NGS assemblies of 16 laboratory
mouse strains into sets of complete chromosomes, leaving < 5% of
sequence unlocalized per set. Various benchmarks, including PCR
testing and realigning of long Pacific Biosciences (PacBio) reads,
suggest only a small number of structural errors in the final
assemblies, comparable with direct assembly approaches. We applied
Ragout 2 to the Mus caroli and Mus pahari genomes, which exhibit
karyotype-scale variations compared with other genomes from the
Muridae family. Chromosome painting maps confirmed most large-scale
rearrangements that Ragout 2 detected. We applied Ragout 2 to
improve draft sequences of three ape genomes that have recently been
published. Ragout 2 transformed three sets of contigs (generated
using PacBio reads only) into chromosome-scale assemblies with
accuracy comparable to chromosome assemblies generated in the
original study using BioNano maps, Hi-C, BAC clones, and FISH.
Genome research. October 19, 2018; doi: 10.1101/gr.236273.118
The Pharmacogenomics of Bipolar Disorder study (PGBD):
identification of genes for lithium response in a prospective sample
Background: Bipolar disorder is a serious and common psychiatric
disorder characterized by manic and depressive mood switches and a
relapsing and remitting course. The cornerstone of clinical
management is stabilization and prophylaxis using mood-stabilizing
medications to reduce both manic and depressive symptoms. Lithium
remains the gold standard of treatment with the strongest data for
both efficacy and suicide prevention. However, many patients do not
respond to this medication, and clinically there is a great need for
tools to aid the clinician in selecting the correct treatment. Large
genome wide association studies (GWAS) investigating retrospectively
the effect of lithium response are in the pipeline; however, few
large prospective studies on genetic predictors to of lithium
response have yet been conducted. The purpose of this project is to
identify genes that are associated with lithium response in a large
prospective cohort of bipolar patients and to better understand the
mechanism of action of lithium and the variation in the genome that
influences clinical response.
Methods/design: This study is an 11-site prospective non-randomized open trial of lithium designed to ascertain a cohort of 700 subjects with bipolar I disorder who experience protocol-defined relapse prevention as a result of treatment with lithium monotherapy. All patients will be diagnosed using the Diagnostic Interview for Genetic Studies (DIGS) and will then enter a 2-year follow-up period on lithium monotherapy if and when they exhibit a score of 1 (normal, not ill), 2 (minimally ill) or 3 (mildly ill) on the Clinical Global Impressions of Severity Scale for Bipolar Disorder (CGI-S-BP Overall Bipolar Illness) for 4 of the 5 preceding weeks. Lithium will be titrated as clinically appropriate, not to exceed serum levels of 1.2 mEq/L. The sample will be evaluated longitudinally using a wide range of clinical scales, cognitive assessments and laboratory tests. On relapse, patients will be discontinued or crossed-over to treatment with valproic acid (VPA) or treatment as usual (TAU). Relapse is defined as a DSM-IV manic, major depressive or mixed episode or if the treating physician decides a change in medication is clinically necessary. The sample will be genotyped for GWAS. The outcome for lithium response will be analyzed as a time to event, where the event is defined as clinical relapse, using a Cox Proportional Hazards model. Positive single nucleotide polymorphisms (SNPs) from past genetic retrospective studies of lithium response, the Consortium on Lithium Genetics (ConLiGen), will be tested in this prospective study sample; a meta-analysis of these samples will then be performed. Finally, neurons will be derived from pluripotent stem cells from lithium responders and non-responders and tested in vivo for response to lithium by gene expression studies. SNPs in genes identified in these cellular studies will also be tested for association to response.
Discussion: Lithium is an extraordinarily important therapeutic drug in the clinical management of patients suffering from bipolar disorder. However, a significant proportion of patients, 30-40 %, fail to respond, and there is currently no method to identify the good lithium responders before initiation of treatment. Converging evidence suggests that genetic factors play a strong role in the variation of response to lithium, but only a few genes have been tested and the samples have largely been retrospective or quite small. The current study will collect an entirely unique sample of 700 patients with bipolar disorder to be stabilized on lithium monotherapy and followed for up to 2 years. This study will produce useful information to improve the understanding of the mechanism of action of lithium and will add to the development of a method to predict individual response to lithium, thereby accelerating recovery and reducing suffering and cost.
BMC Psychiatry. 2016 May 5;16:129; doi: 10.1186/s12888-016-0732-x
Methods/design: This study is an 11-site prospective non-randomized open trial of lithium designed to ascertain a cohort of 700 subjects with bipolar I disorder who experience protocol-defined relapse prevention as a result of treatment with lithium monotherapy. All patients will be diagnosed using the Diagnostic Interview for Genetic Studies (DIGS) and will then enter a 2-year follow-up period on lithium monotherapy if and when they exhibit a score of 1 (normal, not ill), 2 (minimally ill) or 3 (mildly ill) on the Clinical Global Impressions of Severity Scale for Bipolar Disorder (CGI-S-BP Overall Bipolar Illness) for 4 of the 5 preceding weeks. Lithium will be titrated as clinically appropriate, not to exceed serum levels of 1.2 mEq/L. The sample will be evaluated longitudinally using a wide range of clinical scales, cognitive assessments and laboratory tests. On relapse, patients will be discontinued or crossed-over to treatment with valproic acid (VPA) or treatment as usual (TAU). Relapse is defined as a DSM-IV manic, major depressive or mixed episode or if the treating physician decides a change in medication is clinically necessary. The sample will be genotyped for GWAS. The outcome for lithium response will be analyzed as a time to event, where the event is defined as clinical relapse, using a Cox Proportional Hazards model. Positive single nucleotide polymorphisms (SNPs) from past genetic retrospective studies of lithium response, the Consortium on Lithium Genetics (ConLiGen), will be tested in this prospective study sample; a meta-analysis of these samples will then be performed. Finally, neurons will be derived from pluripotent stem cells from lithium responders and non-responders and tested in vivo for response to lithium by gene expression studies. SNPs in genes identified in these cellular studies will also be tested for association to response.
Discussion: Lithium is an extraordinarily important therapeutic drug in the clinical management of patients suffering from bipolar disorder. However, a significant proportion of patients, 30-40 %, fail to respond, and there is currently no method to identify the good lithium responders before initiation of treatment. Converging evidence suggests that genetic factors play a strong role in the variation of response to lithium, but only a few genes have been tested and the samples have largely been retrospective or quite small. The current study will collect an entirely unique sample of 700 patients with bipolar disorder to be stabilized on lithium monotherapy and followed for up to 2 years. This study will produce useful information to improve the understanding of the mechanism of action of lithium and will add to the development of a method to predict individual response to lithium, thereby accelerating recovery and reducing suffering and cost.
Pagerank based clustering of hypertext document collections
Clustering hypertext document collection is an important task in
Information Retrieval. Most clustering methods are based on document
content and do not take into account the hyper-text links. Here we
propose a novel PageRank based clustering (PRC) algorithm which uses
the hypertext structure. The PRC algorithm produces graph
partitioning with high modularity and coverage. The comparison of
the PRC algorithm with two content based clustering algorithms shows
that there is a good match between PRC clustering and content based
clustering.
doi: 10.1145/1390334.1390549
Cerulean: a hybrid assembly using high throughput short and long
reads
Genome assembly using high throughput data with short reads,
arguably, remains an unresolvable task in repetitive genomes, since
when the length of a repeat exceeds the read length, it becomes
difficult to unambiguously connect the flanking regions. The
emergence of third generation sequencing (Pacific Biosciences) with
long reads enables the opportunity to resolve complicated repeats
that could not be resolved by the short read data. However, these
long reads have high error rate and it is an uphill task to assemble
the genome without using additional high quality short reads.
Recently, Koren et al. 2012 [1] proposed an approach to use high
quality short reads data to correct these long reads and, thus, make
the assembly from long reads possible. However, due to the large
size of both dataset (short and long reads), error-correction of
these long reads requires excessively high computational resources,
even on small bacterial genomes. In this work, instead of error
correction of long reads, we first assemble the short reads and
later map these long reads on the assembly graph to resolve repeats.
Lecture Notes in Computer Science(), vol 8126. Springer, Berlin,
Heidelberg; doi: 10.1007/978-3-642-40453-5_27